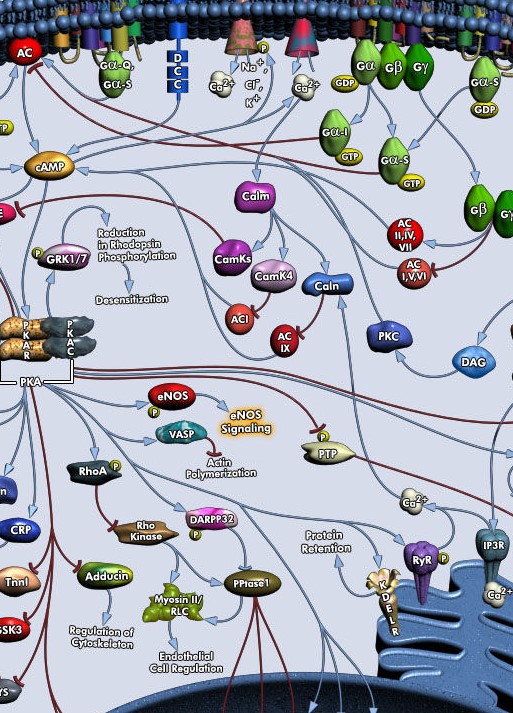
A conjecture based on findings of plasticity in ion channel expression is that the level of expression of various ion channels reflects a memory of the cell. This means that we will have networks of neurons with slightly varying dendritic ion channel populations, which influence not only their general intrinsic excitability, but very specifically influence synaptic transmission through their position at synapses. Some channels aid with transmission, others block or reduce transmission, a phenomenon which has been studied as short-term facilitation or depression. Furthermore, ion channels at the synapse have an influence on synaptic plasticity, again, supporting or blocking plastic changes at the synapse.
This makes a model of neural plasticity more complex than synaptic input-dependent LTP/LTD. A single neuron would need variables at synaptic positions for AMPA and NMDA but also for the main potassium and calcium channels (K-A, Kir, Sk, HCN, L-Ca). In addition, a single set of intrinsic variables could capture the density of ion channels in dendritic shaft position. These variables would allow to express the neural diversity which is a result of memorization or learning.
Hypothesis: Neural plasticity is not primarily input-dependent, instead it is guided by a neural internal state which reflects network processes of knowledge building.
How would the variables that define a neural network be learned? The intrinsic variables would be set by neuromodulatory activation and the ‘internal state’ of the neuron. The synaptic variables would be set from synaptic activation, from neuromodulatory activation, and also from an ‘internal state‘. (In addition, the significant spill-over from synaptic activation which reaches other synapses via the dendrite should also be modeled.)
Let us assume that synaptic stimulation and neuromodulatory activation are well understood. What is the ’internal state‘ of a neuron?
It has been shown that epigenetic modifications are an important factor in memory. These involve methylation changes in DNA and alterations in histones. Their activation is mediated by protein signaling pathways, encompassing kinases like PKA, PKC, CaMKII, MAPK, and other important protein signaling hubs. Long-term neural plasticity and behavioral memory only happen, when the internal conditions are favorable, many reductions or disruptions of internal processes prevent plasticity and behavioral memory. We do not have the data yet to model these processes in detail. But we may use variables for a neuron‘s internal state which is facilitating or inhibiting plasticity. It is an important theoretical question then to understand the impact of internally-guided plasticity on a neural network, one hypothesis is that it helps with conceptual abstraction and knowledge building.
Hypothesis: Only a few neurons in a target area undergo the permanent, lasting changes underlying long-term memory.
Learning events in rodents lead to epigenetic changes in a targeted area, such as amygdala or hippocampus, but it seems as if there are only a few cells, neurons and non-neurons, involved at a time.These changes begin to appear 30 min after a high-frequency stimulation as in dentate gyrus and last 2-5 hours at least. Some have measured the effects 2 weeks after a learning event. Changes are not widespread, as would be expected in distributed memory, instead they are focal, as if only a few cells suffice to store the memory. It is also possible that the changes are strong only in a focal group of neurons, and present, but much weaker in a more distributed group. Focal learning may be seen as a strategy to build more effective knowledge representations, similar to dropout learning techniques which improve feature representation.