

A typical task in theoretical neuroscience is “modeling”, for instance, building and analysing network models of cerebellar cortex that incorporate the diversity of cell types and synaptic connections observed in this structure. The goal is to better understand the functions of these diverse cell types and connections. Approaches from statistical physics, nonlinear dynamics and machine learning can be used, and models should be “ constrained “ by electrophysiological, transcriptomic and connectomic data.
This sounds all very well and quite innocuous. It’s “state of the art”. So what is wrong with this approach that dominates current computational neuroscience?
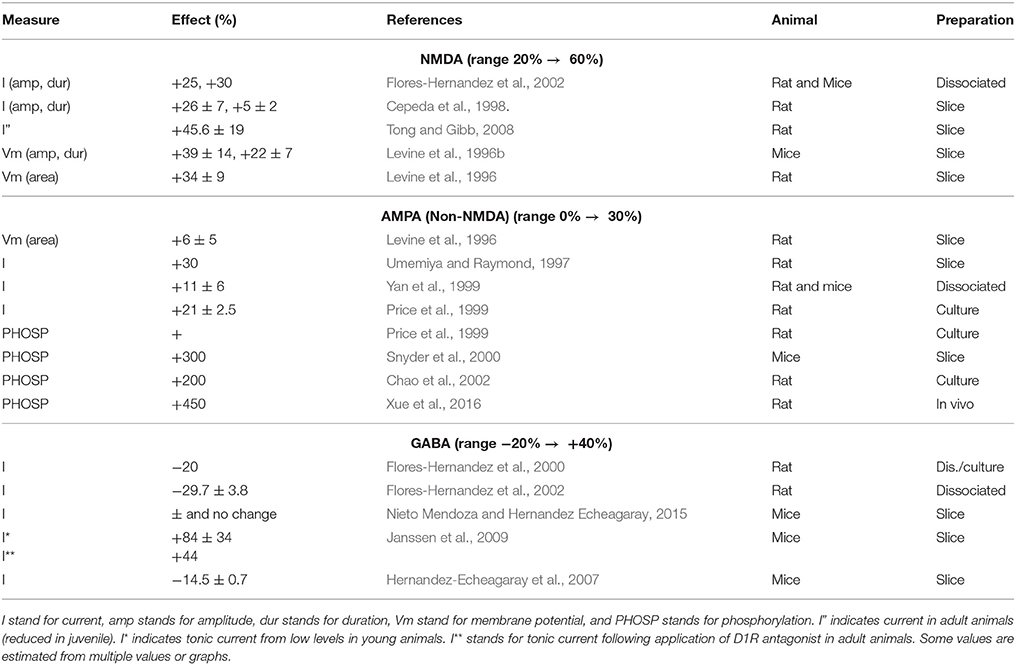
What is wrong is the belief that a detailed, bottom-up model could fulfill the goal of understanding its function. This is only a way to synthesize existing information. Many, most aspects of the model are selected in advance, and much existing information is left out, since it is irrelevant for the model. Irrelevant for the model does not mean that it may not be crucial for the function of the real biological object, such as a neuron. For instance, for decades the fact that many neurons adapt in terms of their ion channels under behavioral learning was simply ignored. Then the notion of whole-neuron learning became acceptable, and under the term “intrinsic excitability” it slowly became part of computational modeling, and now various functions are being discovered, where those changes were first dismissed as “homeostatic” , i.e. only functions in terms of house-keeping were accepted.
If we started a top-down model in terms of what makes sense (what would evolution prefer), and what is logically required or useful for the neuron to operate, then we would have realized a long time ago that whole neuron (intrinsic) excitability is a crucial part of learning. This paper was first published on arxiv 2005, but accepted for publication only in 2013, when intrinsic excitability had become more widely known.






{kind=link}