The feedforward/feedback learning and interaction in the visual system has been analysed as a case of “predictive coding” , the “free energy principle” or “Bayesian perception”. The general principle is very simple, so I will call it “difference learning”. I believe that this is directly comparable (biology doesn’t invent, it re-invents) to what is happening at the cell membrane between external (membrane) and internal (signaling) parameters.
It is about difference modulation: an existing or quiet state, and then new signaling (at the membrane) or by perception (in the case of vision). Now the system has to adapt to the new input. The feedback connections transfer back the old categorization of the new input. This gets added to the perception so that a percept evolves which uses the old categorization together with the new input to achieve quickly an adequate categorization for any perceptual input. There will be a bias of course in favor of existing knowledge, but that makes sense in a behavioral context.
The same thing happens at the membrane. An input signal activates membrane receptors (parameters). The internal parameters – the control structure – transfers back the stored response to the external membrane parameters. And the signal generates a suitable neuronal response according to its effect on external (bottom-up) together with the internal control structure (top-down). The response is now biased in favor of an existing structure, but it also means all signals can quickly be interpreted.
If a signal overcomes a filter, new adaptation and learning of the parameters can happen.
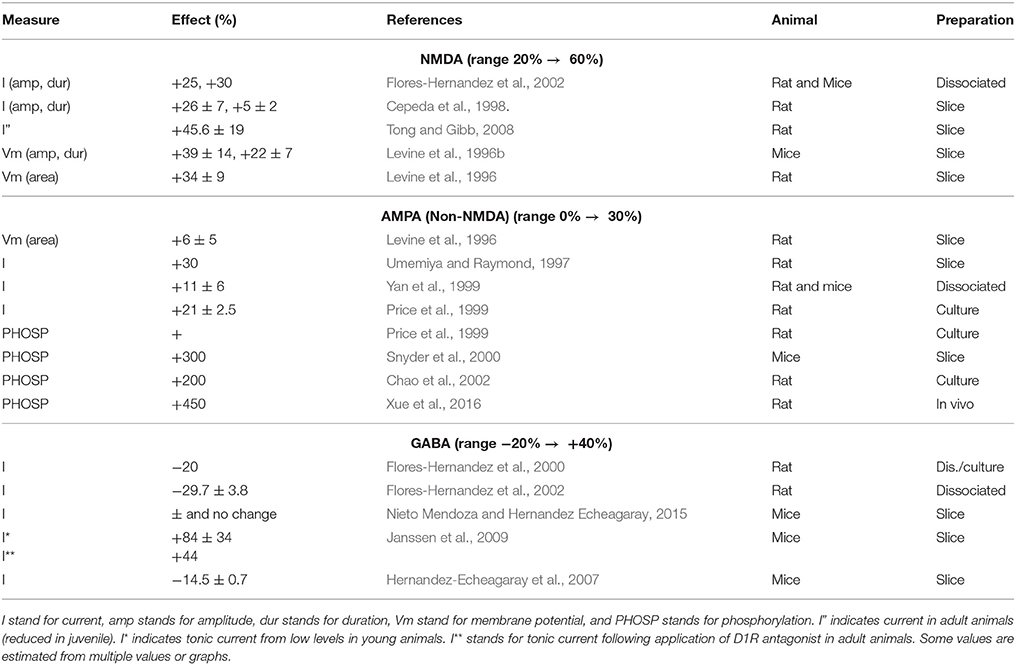
The general principle is difference learning, adaptation on the basis of a difference between encoded information and a new input. This general principle underlies all membrane adaptation, whether at the synapse or the spine, or the dendrite, and all types of receptors, whether AMPA, GABA or GPCR.
We are used to believe that the general principle of neural plasticity is associative learning. This is an entirely different principle and merely derivative of difference learning in certain contexts. Associative learning as the basis of synaptic plasticity goes back more than a 100 years. The idea was that by exposure to different ideas or objects, the connection between them in the mind was strengthened. And it was then conjectured that two neurons (A and B) both of which are activated would strengthen their connection (from A to B). More precisely, as was later often found, A needed to fire earlier than B, in order to encode a sequential relation.
What would be predicted by difference learning? An existing connection would encode the strength of synaptic activation at that site. As long as the actual signal matches, there is no need for adaptation. If it becomes stronger, the synapse may acquire additional receptors by using its internal control structure. This control structure may have requirements about sequentiality. The control structure may also be updated to make the new strength permanent, a new set-point parameter. On the other hand, a weaker than memorized signal will ultimately lead the synapse to wither and die.
Similar outcomes, entirely different principles. Association is encoded by any synapse, and since membrane receptors are plastic, associative learning is a restricted derivative of difference learning.








{kind=link}