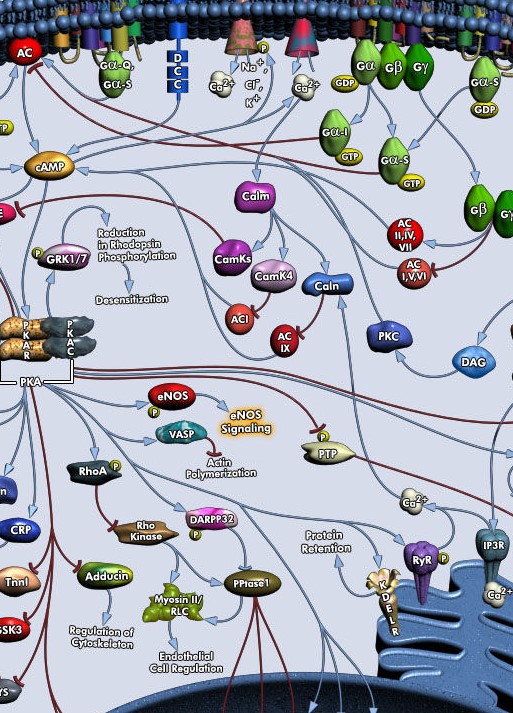
Some time ago, I suggested that equating dopamine with reward learning was a bad idea. Why?
First of all, because it is a myopic view of the role of neuromodulation in the brain, (and also in invertebrate animals). There are at least 4 centrally released neuromodulators, they all act on G-protein-coupled receptors (some not exclusively), and they all have effects on neural processing as well as memory. Furthermore there are myriad neuromodulators which are locally released, and which have similar effects, all acting through different receptors, but on the same internal pathways, activating G-proteins.
Reward learning means that reward increases dopamine release, and that increased dopamine availability will increase synaptic plasticity.
That was always simplistic and like any half-truth misleading.
Any neuromodulator is variable in its release properties. This results from the activity of its NM-producing neurons, such as in locus ceruleus, dorsal raphe, VTA, medulla etc., which receive input, including from each other, and secondly from control of axonal and presynaptic release, which is independent of the central signal. So there is local modulation of release. Given a signal which increases e.g. firing in the VTA, we still need to know which target areas are at the present time responsive, and at which synapses precisely the signal is directed. It depends on the local state of the network, how the global signal is interpreted.
Secondly, the activation of G-protein coupled receptors is definitely an important ingredient in activating the intracellular pathways that are necessary for the expression of plasticity. Roughly, a concurrent activation of calcium and cAMP/PKA (within 10s or so) has been found to be supportive or necessary of inducing synaptic plasticity. However, dopamine, like the other centrally released neuromodulators, acts through antagonistic receptors, increasing or decreasing PKA, increasing or reducing plasticity. It is again local computation which will decide the outcome of NM signaling at each site.
So, is there a take-home message, rivaling the simplicity of dopamine=reward?
NMs alter representations (=thought) and memorize them (=memory) but the interpretation is flexible at local sites (=learn and re-learn).
Dopamine alters thought and memory in a way that can be learned and re-learned.
Back in 1995 I came up with the idea of analysing neuromodulators like dopamine as a method of introducing global parameters into neural networks, which were considered at the time to admit only local, distributed computations. It seemed to me then, as now, that the capacity for global control of huge brain areas (serotonergic, cholinergic, dopaminergic and noradrenergic systems), was really what set neuromodulation apart from the neurotransmitters glutamate and GABA. There is no need to single out dopamine as the one central signal, which induces simple increases in its target areas, when in reality changes happen through antagonistic receptors, and there are many central signals. Also, the concept of hedonistic reward is badly defined and essentially restricted to Pavlovian conditioning for animals and addiction in humans.
Since the only known global parameter in neural networks at the time occurred in reinforcement learning, some people created a match, using dopamine as the missing global reinforcement signal (Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997). That could not work, because reinforcement learning requires proper discounting within a decision tree. But the idea stuck. Ever since I have been upset at this primitive oversimplification. Bad ideas in neuroscience.
Scheler, G and Fellous, J-M: Dopamine modulation of prefrontal delay activity- reverberatory activity and sharpness of tuning curves. Neurocomputing, 2001.
Scheler, G. and Schumann, J: Presynaptic modulation as fast synaptic switching: state-dependent modulation of task performance. Proceedings of the International Joint Conference on Neural Networks 2003, Volume: 1. DOI: 10.1109/IJCNN.2003.1223347